Introduction
Protein structure prediction is a crucial part of understanding the function of proteins in various biological processes. Structural bioinformatics is a field that aims to predict the three-dimensional structures of proteins and to use that information to gain insights into their functions. This article will provide a comprehensive guide to protein structure prediction using structural bioinformatics.

What is Structural Bioinformatics?
Structural bioinformatics is an interdisciplinary field that combines molecular biology, biochemistry, and computer science to predict the three-dimensional structure of proteins. It involves the use of computational algorithms to analyze protein sequences, predict their structure, and simulate protein dynamics.
Why is Protein Structure Important?
The three-dimensional structure of a protein is crucial to understanding its function. Proteins perform a wide range of functions in biological systems, including catalyzing chemical reactions, regulating gene expression, and transporting molecules across cell membranes. The function of a protein is determined by its structure, and any changes in the structure can lead to changes in function.
Protein Structure Prediction Methods
There are several methods for predicting protein structures, each with its advantages and disadvantages. Here are some of the most commonly used methods:
Homology Modeling
Homology modeling is a method for predicting the structure of a protein based on its similarity to a known structure. This method uses a template structure as a guide to predict the structure of the target protein. It is a fast and accurate method when there is a close homolog available.
Ab Initio Methods
Ab initio methods are based on physical principles and do not rely on homology to predict protein structures. These methods use algorithms to sample conformational space and identify low-energy conformations that are likely to be the native state of the protein.
Hybrid Methods
Hybrid methods combine homology modeling and ab initio methods to predict protein structures. These methods use homology modeling to generate an initial model and then refine the model using ab initio methods.
Protein Structure Databases
Protein structure databases are an essential resource for structural bioinformatics. These databases contain information about the three-dimensional structure of proteins, including their atomic coordinates, experimental data, and functional annotations. Here are some of the most commonly used protein structure databases:
Protein Data Bank (PDB)
The Protein Data Bank (PDB) is a database that contains experimentally determined protein structures. It is the most comprehensive and widely used protein structure database, with over 170,000 structures available as of 2021. The database is maintained by the Worldwide Protein Data Bank, a collaboration between organizations in the United States, Europe, and Asia.
The PDB contains information on the three-dimensional structure of proteins, including the positions of atoms, amino acid residues, and ligands. The structures are typically determined using techniques such as X-ray crystallography or nuclear magnetic resonance spectroscopy. Each structure in the PDB is assigned a unique identifier, called a PDB code, which allows researchers to easily access and analyze the structure.
The PDB also includes tools for visualizing and analyzing protein structures, such as the Jmol viewer and the RCSB Ligand Explorer. These tools allow researchers to explore the structure of a protein and analyze its interactions with ligands or other molecules.
The PDB is used in a wide range of applications, including drug discovery, protein engineering, and understanding the structure and function of biological molecules. It is an essential resource for researchers in structural bioinformatics, who use the database to compare and analyze protein structures and to develop computational tools for predicting protein structure and function.
To add a new structure to the PDB, researchers must submit their experimental data to one of the member organizations of the Worldwide Protein Data Bank. The data are then reviewed and processed before being added to the database. The submission process is designed to ensure that the data are accurate and of high quality, and that they conform to the standards set by the PDB.
Once a structure is added to the PDB, it becomes publicly available and can be accessed by researchers around the world. The database is regularly updated with new structures, and older structures are periodically re-analyzed to ensure their accuracy and completeness.
In addition to providing access to experimentally determined protein structures, the PDB also includes tools for predicting protein structure and function. These tools use computational methods to generate models of protein structure based on the amino acid sequence. While these models may not be as accurate as experimentally determined structures, they can still provide valuable insights into protein function and can be used in drug discovery and other applications.
The Protein Data Bank is an essential resource for researchers in structural biology and bioinformatics. Its comprehensive collection of protein structures and tools for analysis and prediction have enabled many important discoveries and advancements in the field.
Structural Classification of Proteins (SCOP)
The Structural Classification of Proteins (SCOP) is a database that organizes proteins based on their structure and function. It uses a hierarchical classification scheme to group related protein structures into categories. The classification system includes four levels: Class, Fold, Superfamily, and Family.
The Class level is the broadest classification and is based on the overall shape and organization of the protein structure. The Fold level is based on the arrangement of secondary structural elements within the protein, while the Superfamily level groups proteins with similar structure and function. Finally, the Family level groups proteins that have highly similar sequences and structure.
The SCOP database includes detailed information about the structure and function of each protein, including annotations, literature references, and images. It also provides tools for visualizing and analyzing protein structures.
The SCOP database is widely used in protein structure research to classify and compare protein structures. It can be used to identify relationships between proteins, predict protein function based on structural similarities, and guide drug discovery efforts. The database is constantly updated with new protein structures and annotations, making it a valuable resource for the scientific community.
CATH
CATH (Class, Architecture, Topology, Homology) is a database that classifies protein structures based on their architecture, topology, and homology. The database was first developed in the early 1990s by Christine Orengo and her colleagues at University College London, and it has since become an important resource for structural bioinformatics research.
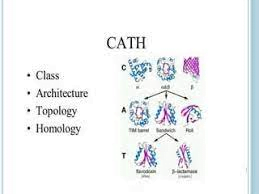
The CATH database contains information on the three-dimensional structures of proteins and classifies them into four main categories based on their structure and function. The first category is Class, which defines the main protein fold type, such as alpha, beta, or alpha-beta. The second category is Architecture, which describes the overall shape of the protein and its arrangement of secondary structure elements. The third category is Topology, which specifies the connectivity of the secondary structure elements. Finally, the Homology category groups proteins based on their evolutionary relationships.
The CATH database is continually updated and currently contains over 300,000 protein structures from the Protein Data Bank (PDB). In addition to the classification of protein structures, the database also provides information on sequence families, functional annotations, and protein-protein interactions.
One of the key applications of the CATH database is in the study of protein evolution and function. By analyzing the structures and homologies of proteins in the database, researchers can gain insights into the evolutionary relationships between proteins and their functions. The database can also be used to identify functionally important regions of proteins and to design experiments to test their function.
Another important application of the CATH database is in the field of drug discovery. By understanding the structure and function of proteins, researchers can design drugs that specifically target them. The CATH database can be used to identify potential drug targets and to optimize the design of drug molecules for maximum efficacy.
The CATH database is a valuable resource for structural bioinformatics research, providing a comprehensive classification of protein structures based on their architecture, topology, and homology. Its applications include the study of protein evolution and function, drug discovery, and protein engineering.

Applications of Protein Structure Prediction
Protein structure prediction has several applications in molecular biology, biochemistry, and drug discovery. Here are some of the most important applications:
Drug Discovery
Protein structure prediction is an essential tool in drug discovery. It allows researchers to design drugs that target specific proteins and to optimize the drug’s properties for maximum efficacy.
Protein Engineering
Protein structure prediction can be used to engineer proteins with specific functions. By modifying the amino acid sequence of a protein, researchers can change its structure and function.
Understanding Protein Function
Protein structure prediction can provide insights into the function of proteins. By comparing the structures of different proteins, researchers can identify common structural motifs and determine their functional significance.
Conclusion
Structural bioinformatics is an essential field in molecular biology and biochemistry. Protein structure prediction plays a vital
role in understanding protein function and is crucial in drug discovery and protein engineering. There are several methods for predicting protein structures, including homology modeling, ab initio methods, and hybrid methods. Protein structure databases such as the Protein Data Bank, SCOP, and CATH are invaluable resources for structural bioinformatics research.
In summary, structural bioinformatics has revolutionized the field of molecular biology and biochemistry by allowing researchers to predict and analyze protein structures. As the field continues to evolve, we can expect even more exciting discoveries and applications of protein structure prediction.
FAQs
- What is the difference between homology modeling and ab initio methods?
- Homology modeling relies on a known protein structure to predict the structure of a similar protein, while ab initio methods predict the structure based on physical principles without relying on a known structure.
- What are some of the most commonly used protein structure databases?
- The most commonly used protein structure databases include the Protein Data Bank, SCOP, and CATH.
- How is protein structure prediction used in drug discovery?
- Protein structure prediction is used to design drugs that target specific proteins and to optimize the drug’s properties for maximum efficacy.
- Protein structure prediction is used to design drugs that target specific proteins and to optimize the drug’s properties for maximum efficacy.
- What are some applications of protein engineering? Protein engineering can be used to create proteins with specific functions, such as enzymes that catalyze specific reactions or antibodies that recognize specific molecules.
- What is the importance of understanding protein structure?
- The three-dimensional structure of a protein is crucial to understanding its function. By understanding the structure of a protein, researchers can gain insights into its function and develop new drugs and therapies.